






最新ニュース
EU 11/17付け、医療機器指令 MDDの整合規格が更新されました
MDD医療機器指令への適合宣言において、基本要件適合への適合を示す目的として実質的に強制力を持つ欧州整合規格()のリストが更新されました。
(MEDICAの展示会最終日の翌日に更新となりました)
MDD 整合規格 Harmonized Standardのリンクはこちら
EN ISO13485:2016/AC:2016が2019年の3月末よりEN ISO13485:2012に置き換わります。
|
CEN |
EN ISO 13485:2016 (new) Medical devices - Quality management systems - Requirements for regulatory purposes (ISO 13485:2016) |
EN ISO 13485:2012 |
31/03/2019 |
|
|
EN ISO 13485:2016/AC:2016 (new) |
また、ラベル、ラベリングや提供される情報に使用されるシンボルの規格として長年使用されてきたEN980:2008も年末をもってEN ISO15223-1:2016に置き換わります。
年明けにMDD監査を迎えられる企業様は適合宣言書の更新もお忘れなく。
|
CEN |
EN ISO 15223-1:2016 (new) Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements (ISO 15223-1:2016, Corrected version 2017-03) |
EN 980:2008 |
31/12/2017 |
また、60601-1シリーズでは医療診断用のMR装置の規格、-2-33が新しくなっています。
EN 60601-2-33:2010 Part 2-33: Particular requirements for the basic safety and essential performance of magnetic resonance equipment for medical diagnosis
EU 11/17付け、体外診断用医療機器指令IVDDの整合規格が更新されました
IVDD 体外診断用医療機器指令への適合宣言において、基本要件適合への適合を示す目的として実質的に強制力を持つ欧州整合規格のリストが更新されました。
(MEDICAの展示会最終日の翌日に更新となりました)
IVDD 整合規格 Harmonized Standardのリンクはこちら
EN ISO13485:2016/AC:2016が2019年の3月末よりEN ISO13485:2012に置き換わります。
|
CEN |
EN ISO 13485:2016 (new) Medical devices - Quality management systems - Requirements for regulatory purposes (ISO 13485:2016) |
EN ISO 13485:2012Note 2.1 |
31/03/2019 |
|
|
EN ISO 13485:2016/AC:2016 (new) |
また、ラベル、ラベリングや提供される情報に使用されるシンボルの規格として長年使用されてきたEN980:2008も年末をもってEN ISO15223-1:2016に置き換わります。
|
CEN |
EN ISO 15223-1:2016 (new) Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements (ISO 15223-1:2016, Corrected version 2017-03) |
EN 980:2008Note 2.1 |
31/12/2017 |
また、体外診断用医療機器の個別規格となる61010-2-101はまだ2002年度版のままです。IEC/EN61010-1第三版の適用判断についてはまだ法的製造業者様の方針に委ねられています。
|
Cenelec |
EN 61010-2-101:2002 Safety requirements for electrical equipment for measurement, control, and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment |
17/12/2002 |
台湾 TFDAへのQSD、製品登録(PR)申請窓口(画像)
本日も弊社台北本社の申請スタッフがTFDAを訪問しています。
(台北は快晴、30℃近い気温となっているようです)
QSDや製品の登録申請時、また、申請後のTFDAの審査担当の方との打ち合わせでTFDAを訪問いたします。
申請後は電話での確認が多いですが、直接書類を確認しながらの説明が重要な場面は沢山ございます。
下記はTFDAの外観、受付エリアの画像となります。





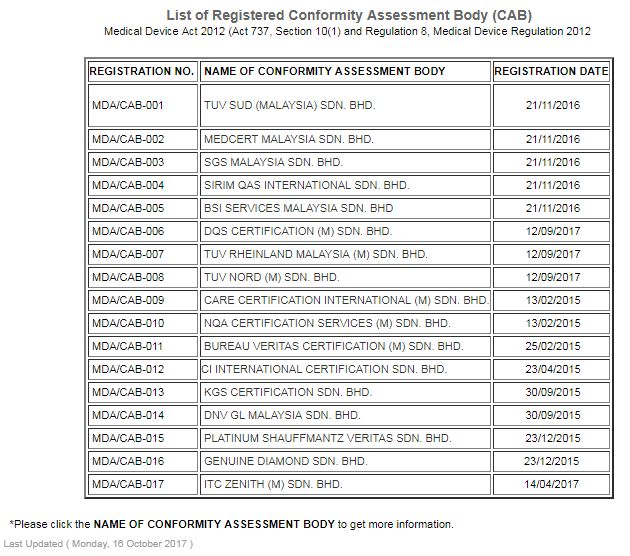
マレーシア Conformity Assessment Body (CAB)適合性評価機関
下記は2017年10月16日時点での最新のCABリストとなります。
合計17のCABがマレーシアMDAに登録されており、CAB毎に審査可能な製品が異なります。
日本では民間の登録認証機関が認証できる認証範囲を管理医療機器では体外診断用医薬品を含む22区分、高度管理医療機器では11区分が一覧にまとめられています。
http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000074863.html
最多の審査範囲を持つCABはDQS CERTIFICATION (M) SDN.BHD.です。
DQS社の特徴として、能動・非能動機器を広くカバーし、ソフトウェアや能動埋込み機器まで登録範囲に持っています。
※マレーシア MDAの最新CABリストはこちら
弊社のマレーシア・オフィスは複数のCABへの申請実績がございます。
CABとのコミュニケーション以上に重要となる申請資料の作成に力を入れ、審査完了までの時間短縮を実現しています。
また、マレーシアMDA(医療機器庁)とも蜜にコミュニケーションを行い、製品登録のみならず規制調査業務でもより精度の高い情報提供が可能です。
ベトナム Decree No. 36/2016/ND-CPのラベル要求について
ベトナムでの医療機器規制では、これまでCircular 24/2011/TT-BYT の要求を満たす必要がありました。
Circular 24/2011/TT-BYT では、ラベルの要求は第3条の4.で以下のように規定されていました。
|
4. Labeling of the imported medical device: It must be implemented in accordance with the regulations of the Government’s Decree No. 89/2006/ND-CP dated 30 August 2006 on labeling of goods and Circular No. 09/2007/TT-BKHCN dated 6 April 2007 of Ministry of Science and Technology guiding the implementation of a number of articles of the Government’s Decree No. 89/2006/ND-CP dated 30 August 2006 on labeling of goods and several related regulations. |
※2006年8月30日付のDecree No. 89/2006、2997年4月6日付のCircular No. 09/2007に従うこと。
今回導入されたDecree No.36/2016では、第54条で規定されています。
|
Article 54. Labels of medical equipment 1. The labeling of medical equipment shall comply with regulations in Decree No. 89/2006/ND-CP dated August 30, 2006 by the Government on labeling of goods and the label must contain: a) Name of the medical equipment; b) The registration number of free sale of the medical equipment; c) Name and address of the holder of the registration number of free sale of the medical equipment; d) The origin of the medical equipment; dd) Date of production or expiry date. The date of production and the expiry date must be written in the format [dd/mm/yyyy] or [mm/yyyy]. e) Number of batch or the seri number of the medical equipment; g) Guidance for seeking information about the warranty provider, guidlines for using the medical equipment, technical documents serving the repair and maintenance according to regulations in clause 2 Article 17 of this Decree. 2. Medical equipment imported into Vietnam whose label does not contain sufficiently the information specified in clause 1 of this Article must be enclosed with a supplementary label containing such information written in Vietnamese and the original label of the equipment shall be retained. |
<参考訳>
1.2006年8月30日付のDecree No. 89/2006に従い、ラベルは以下も満たす事:
a)製品名称
b)医療機器の登録番号:Registration Number 新しく追加
c)医療機器の自由販売に責任を持つ組織名と住所
d)医療機器の原産地
dd)製造日又は使用期限: 記載方法は[dd/mm/yyyy] 又は [mm/yyyy]に従うこと。
e)医療機器のバッチナンバー又はシリアルナンバー
g)保証、機器の使用、2項17条の要求を満たす修理・保守の提供者情報
2.ベトナムに輸入される医療機器で上記1項の要求事項を満足しないラベルについては、元のラベルと一緒にベトナム語の付属ラベルで対応つけること。
マレーシア MDAは 非矯正コンタクトレンズを2018年1月1日より医療機器として取り扱う
マレーシアの医療機器規制当局 MDA(Medical Device Authority)は非矯正コンタクトレンズを医療機器として取り扱うことを決定しました。
登録に関する重要なポイントは下記の2点となります。
1)強制日までに輸入・輸出・市場へ出荷した事業者は強制日から6ヶ月以内に登録申請をすること
2)非矯正コンタクトレンズの登録申請を実施した事業者は輸入・輸出・市場への出荷を継続できること
※マレーシアMDA当局のリンクはこちら
(英語の本文は4ページ目以降です)
※ご参考まで、アメリカFDAのDecorative, Non-correntive Contact Lensに関する文書のリンクはこちら
<更新>医療機器規制勉強会 2017年12月15日(金)OMETA様主催勉強会のご案内
OMETA医療機器規則勉強会2017 大阪会場のプログラム内容がアップデートされました。
※UDI規則の追加
20171215OMETA海外医療機器規制勉強会_大阪会場ver2.pdf
<プログラム>(予定)
14:00-14:20 AHWP (Asian Harmonization Working Party)年次総会2017 参加報告
14:20-14:30 UDI規則に関する各国アップデート
14:30-15:30 ASEAN諸国の規制アップデート
15:40-16:20 インドネシア製品登録の状況について
台湾 TFDA クラス分類ルールの更新について
台湾の薬事申請、製品登録ではTFDAが発表するクラス分類のルールを定期的に確認することが大切です。
クラスアップ品についてはQSD申請が必要になったり、製品登録のスケジュールや販売計画の大幅な見直し、現地代理店との契約の見直し等が必要になります。
また、TFDAの規則2016(2016 rule)では新しい製品の追加情報も確認が出来ます。
<新規追加品の例>
I.0008 經皮皮膚刺激器 Transcutaneous skin stimulator
※”化妝水導入“が主たる目的であれば医療機器ではない
上記のように医療機器か非医療機器かの判断も重要なポイントとなります。
クアルテックでは、アジア 7拠点のネットーワークで最新のクラス分類の調査も対応しております。
お気軽にお問い合わせください。
台湾 政府の新南向政策に対しTFDAは医療機器の会議を開催
台湾政府の新南向政策推進計画に対し、去る10月12日にTFDAは医療機器規制の情報交換を目的として会議を開催いたしました。
弊社の本社スタッフもセミナーに参加し、アセアンMDD(ASEAN MDD)やEU、インド、マレーシアの最新情報を入手いたしました。
新南政策のリンク↓
http://www.roc-taiwan.org/jp_ja/post/40085.html
"Conference on Medical Device Regulations of Europe and Asia"
トピック:
-ASEAN Medical Device Directive (AMDD)
-New EU Regulations on Meidal Devices and First Steps of their implementation
-Regulation of Medical Device and In vitro Diagnostics in India
-Medical Device Registration Requirements in Malaysia
セミナーでの最新情報は12月のOMETA様のセミナーでも情報共有させていただきます。
医療機器規制勉強会 2017年12月15日(金)OMETA様主催勉強会のご案内
来る12月15日(金)の午後2時より特定非営利活動法人 海外医療機器技術協力会(OMETA)主催の勉強会が大阪のドーンセンターにて開催されます。ASEANの規制とインドネシア製品登録についてお話させていただきます。
詳細は下記のプログラム資料をご確認下さい。