









アジア・ASEAN各国の規制対応で規制情報がクリアな国にシンガポールがあります。
シンガポールへの製品登録の第一ステップとして、規制当局のHSAに対しクラス分類の確認からスタートします。
医療機器に対するクラス分類は、そのガイダンス文書を確認すると欧州のクラス分類とほぼ同じものであることがわかります。
例えば医療機器のクラス分類のルールはシンガポール:16、EU:18となります。
ルールの数こそ違えど、その内容は全く同じか英語の表現が異なる程度の違いです。
弊社でもクラス分類の確認相談をいただいた場合には「欧州ではどのクラスですか?」とまずはお聞きしています。
下記までクラス分類に関連する情報をまとめてみましたので参考にしてください。
シンガポールの製品登録、現地代理人はクアルテックにお任せください。
国内外の医療機器メーカー様の現地代理人として、製品登録から市販後管理まで、貴社の薬事部門のアウトソース先として選ばれています。
MDD医療機器指令への適合宣言において、基本要件適合への適合を示す目的として実質的に強制力を持つ欧州整合規格()のリストが更新されました。
(MEDICAの展示会最終日の翌日に更新となりました)
MDD 整合規格 Harmonized Standardのリンクはこちら
EN ISO13485:2016/AC:2016が2019年の3月末よりEN ISO13485:2012に置き換わります。
|
CEN |
EN ISO 13485:2016 (new) Medical devices - Quality management systems - Requirements for regulatory purposes (ISO 13485:2016) |
EN ISO 13485:2012 |
31/03/2019 |
|
|
EN ISO 13485:2016/AC:2016 (new) |
また、ラベル、ラベリングや提供される情報に使用されるシンボルの規格として長年使用されてきたEN980:2008も年末をもってEN ISO15223-1:2016に置き換わります。
年明けにMDD監査を迎えられる企業様は適合宣言書の更新もお忘れなく。
|
CEN |
EN ISO 15223-1:2016 (new) Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements (ISO 15223-1:2016, Corrected version 2017-03) |
EN 980:2008 |
31/12/2017 |
また、60601-1シリーズでは医療診断用のMR装置の規格、-2-33が新しくなっています。
EN 60601-2-33:2010 Part 2-33: Particular requirements for the basic safety and essential performance of magnetic resonance equipment for medical diagnosis
IVDD 体外診断用医療機器指令への適合宣言において、基本要件適合への適合を示す目的として実質的に強制力を持つ欧州整合規格のリストが更新されました。
(MEDICAの展示会最終日の翌日に更新となりました)
IVDD 整合規格 Harmonized Standardのリンクはこちら
EN ISO13485:2016/AC:2016が2019年の3月末よりEN ISO13485:2012に置き換わります。
|
CEN |
EN ISO 13485:2016 (new) Medical devices - Quality management systems - Requirements for regulatory purposes (ISO 13485:2016) |
EN ISO 13485:2012Note 2.1 |
31/03/2019 |
|
|
EN ISO 13485:2016/AC:2016 (new) |
また、ラベル、ラベリングや提供される情報に使用されるシンボルの規格として長年使用されてきたEN980:2008も年末をもってEN ISO15223-1:2016に置き換わります。
|
CEN |
EN ISO 15223-1:2016 (new) Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements (ISO 15223-1:2016, Corrected version 2017-03) |
EN 980:2008Note 2.1 |
31/12/2017 |
また、体外診断用医療機器の個別規格となる61010-2-101はまだ2002年度版のままです。IEC/EN61010-1第三版の適用判断についてはまだ法的製造業者様の方針に委ねられています。
|
Cenelec |
EN 61010-2-101:2002 Safety requirements for electrical equipment for measurement, control, and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment |
17/12/2002 |
本日も弊社台北本社の申請スタッフがTFDAを訪問しています。
(台北は快晴、30℃近い気温となっているようです)
QSDや製品の登録申請時、また、申請後のTFDAの審査担当の方との打ち合わせでTFDAを訪問いたします。
申請後は電話での確認が多いですが、直接書類を確認しながらの説明が重要な場面は沢山ございます。
下記はTFDAの外観、受付エリアの画像となります。





下記は2017年10月16日時点での最新のCABリストとなります。
合計17のCABがマレーシアMDAに登録されており、CAB毎に審査可能な製品が異なります。
日本では民間の登録認証機関が認証できる認証範囲を管理医療機器では体外診断用医薬品を含む22区分、高度管理医療機器では11区分が一覧にまとめられています。
http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000074863.html
最多の審査範囲を持つCABはDQS CERTIFICATION (M) SDN.BHD.です。
DQS社の特徴として、能動・非能動機器を広くカバーし、ソフトウェアや能動埋込み機器まで登録範囲に持っています。
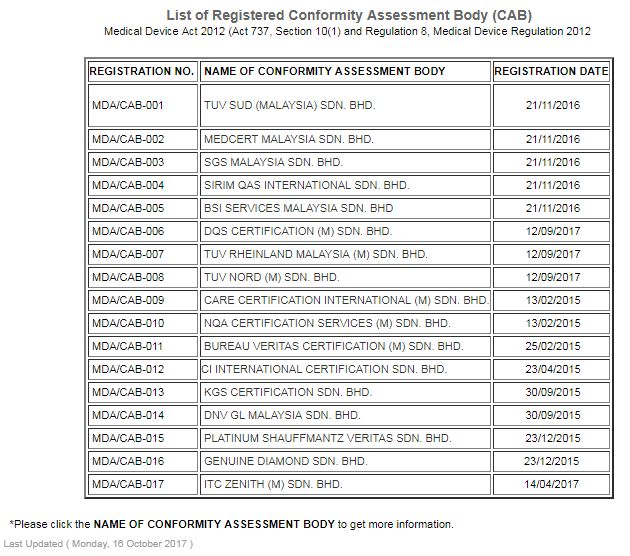
※マレーシア MDAの最新CABリストはこちら
弊社のマレーシア・オフィスは複数のCABへの申請実績がございます。
CABとのコミュニケーション以上に重要となる申請資料の作成に力を入れ、審査完了までの時間短縮を実現しています。
また、マレーシアMDA(医療機器庁)とも蜜にコミュニケーションを行い、製品登録のみならず規制調査業務でもより精度の高い情報提供が可能です。
ベトナムでの医療機器規制では、これまでCircular 24/2011/TT-BYT の要求を満たす必要がありました。
Circular 24/2011/TT-BYT では、ラベルの要求は第3条の4.で以下のように規定されていました。
|
4. Labeling of the imported medical device: It must be implemented in accordance with the regulations of the Government’s Decree No. 89/2006/ND-CP dated 30 August 2006 on labeling of goods and Circular No. 09/2007/TT-BKHCN dated 6 April 2007 of Ministry of Science and Technology guiding the implementation of a number of articles of the Government’s Decree No. 89/2006/ND-CP dated 30 August 2006 on labeling of goods and several related regulations. |
※2006年8月30日付のDecree No. 89/2006、2997年4月6日付のCircular No. 09/2007に従うこと。
今回導入されたDecree No.36/2016では、第54条で規定されています。
|
Article 54. Labels of medical equipment 1. The labeling of medical equipment shall comply with regulations in Decree No. 89/2006/ND-CP dated August 30, 2006 by the Government on labeling of goods and the label must contain: a) Name of the medical equipment; b) The registration number of free sale of the medical equipment; c) Name and address of the holder of the registration number of free sale of the medical equipment; d) The origin of the medical equipment; dd) Date of production or expiry date. The date of production and the expiry date must be written in the format [dd/mm/yyyy] or [mm/yyyy]. e) Number of batch or the seri number of the medical equipment; g) Guidance for seeking information about the warranty provider, guidlines for using the medical equipment, technical documents serving the repair and maintenance according to regulations in clause 2 Article 17 of this Decree. 2. Medical equipment imported into Vietnam whose label does not contain sufficiently the information specified in clause 1 of this Article must be enclosed with a supplementary label containing such information written in Vietnamese and the original label of the equipment shall be retained. |
<参考訳>
1.2006年8月30日付のDecree No. 89/2006に従い、ラベルは以下も満たす事:
a)製品名称
b)医療機器の登録番号:Registration Number 新しく追加
c)医療機器の自由販売に責任を持つ組織名と住所
d)医療機器の原産地
dd)製造日又は使用期限: 記載方法は[dd/mm/yyyy] 又は [mm/yyyy]に従うこと。
e)医療機器のバッチナンバー又はシリアルナンバー
g)保証、機器の使用、2項17条の要求を満たす修理・保守の提供者情報
2.ベトナムに輸入される医療機器で上記1項の要求事項を満足しないラベルについては、元のラベルと一緒にベトナム語の付属ラベルで対応つけること。
マレーシアの医療機器規制当局 MDA(Medical Device Authority)は非矯正コンタクトレンズを医療機器として取り扱うことを決定しました。
登録に関する重要なポイントは下記の2点となります。
1)強制日までに輸入・輸出・市場へ出荷した事業者は強制日から6ヶ月以内に登録申請をすること
2)非矯正コンタクトレンズの登録申請を実施した事業者は輸入・輸出・市場への出荷を継続できること
※マレーシアMDA当局のリンクはこちら
(英語の本文は4ページ目以降です)
※ご参考まで、アメリカFDAのDecorative, Non-correntive Contact Lensに関する文書のリンクはこちら
OMETA医療機器規則勉強会2017 大阪会場のプログラム内容がアップデートされました。
※UDI規則の追加
20171215OMETA海外医療機器規制勉強会_大阪会場ver2.pdf
<プログラム>(予定)
14:00-14:20 AHWP (Asian Harmonization Working Party)年次総会2017 参加報告
14:20-14:30 UDI規則に関する各国アップデート
14:30-15:30 ASEAN諸国の規制アップデート
15:40-16:20 インドネシア製品登録の状況について
台湾の薬事申請、製品登録ではTFDAが発表するクラス分類のルールを定期的に確認することが大切です。
クラスアップ品についてはQSD申請が必要になったり、製品登録のスケジュールや販売計画の大幅な見直し、現地代理店との契約の見直し等が必要になります。
また、TFDAの規則2016(2016 rule)では新しい製品の追加情報も確認が出来ます。
<新規追加品の例>
I.0008 經皮皮膚刺激器 Transcutaneous skin stimulator
※”化妝水導入“が主たる目的であれば医療機器ではない
上記のように医療機器か非医療機器かの判断も重要なポイントとなります。
クアルテックでは、アジア 7拠点のネットーワークで最新のクラス分類の調査も対応しております。
お気軽にお問い合わせください。
台湾政府の新南向政策推進計画に対し、去る10月12日にTFDAは医療機器規制の情報交換を目的として会議を開催いたしました。
弊社の本社スタッフもセミナーに参加し、アセアンMDD(ASEAN MDD)やEU、インド、マレーシアの最新情報を入手いたしました。
新南政策のリンク↓
http://www.roc-taiwan.org/jp_ja/post/40085.html
"Conference on Medical Device Regulations of Europe and Asia"
トピック:
-ASEAN Medical Device Directive (AMDD)
-New EU Regulations on Meidal Devices and First Steps of their implementation
-Regulation of Medical Device and In vitro Diagnostics in India
-Medical Device Registration Requirements in Malaysia
セミナーでの最新情報は12月のOMETA様のセミナーでも情報共有させていただきます。
来る12月15日(金)の午後2時より特定非営利活動法人 海外医療機器技術協力会(OMETA)主催の勉強会が大阪のドーンセンターにて開催されます。ASEANの規制とインドネシア製品登録についてお話させていただきます。
詳細は下記のプログラム資料をご確認下さい。
国内の一変や軽微変更のように、マレーシアでは変更申請のガイダンス文書第一版が今年の1月に発表されています。
この度、第二版が10月にMDA/GD/0020として発表されました。
マレーシアに限らず、医療機器の変更管理は登録を正しく維持していく上で非常に重要なタスクとなります。
ガイダンス文書は↓
Change Notification for Registered Medical Device
新規申請が必要となるカテゴリ1の変更、変更の実施と市場への出荷前にMDAへの申請と許可を要するカテゴリ2、そして当局の受け入れ後直ぐに実施出来るカテゴリ3の3カテゴリに分類されています。
a) Category 1 changes of medical devices that affect their safety and performance and require new registration of the medical device;
b) Category 2 changes are changes that require evaluation and endorsement from the MDA prior to implementation of the change and before placing in the market; and
c) Category 3 changes may be implemented immediately upon receipt of the acknowledgement from the Authority.
本文では具体的な変更事例と必要書類、附属書Eの中で変更申請のフローチャートによる解説など、見やすい内容になっています。
クラルテック・グループはマレーシアのみならずアジア・ASEAN7カ国の薬事登録を専門としています。
EUのAnnexIIのような自己認証とは違う管理が求められますが、必要に応じて各規制当局へ変更申請の要否判断も対応しておりますので、判断に迷われた場合には、お気軽にご相談ください。
欧州 CEマーク対応中の企業様は、まだ先のようで直ぐにやってくるMDRやIVDR対応は悩みのタネかもしれません。
これはノーティファイドボディのみならず、欧州代理人も同じ状況です。
その一つとして、新規則に盛り込まれたDefective Devices (欠陥品・不良品)に対する欧州代理人の賠償責任の負担があります。
Given that pivotal role, for the purposes of enforcement it is appropriate to make the authorised representative legally liable for defective devices in the event that a manufacturer established outside the Union has not complied with its general obligations.
The liability of the authorised representative provided for in this regulation is without prejudice to the provisions of Directive 85/374/EEC, and accordingly the authorised representative should be jointly and severally liable with the importer and the manufacturer.
http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=OJ:L:2017:117:FULL&from=EN
原文(34)より。
先般の大きな訴訟問題も関係しているかもしれませんが、例えば訴訟大国と言われるアメリカでは輸入業者、販売代理店などもPL賠償責任保険に加入するのは一般的です。
現地販売代理店は設計責任や製造責任はありませんが、流通責任に対し訴えを起こされたら必ず応訴しなければなりません。
その訴訟に対する(備える)保険となります。
欧州代理人もMDR/IVDR対応に向け保険会社と面談を行っています。
メーカーに比べ保険料率は低いと言えど、金銭的な負担は代理人費用の増加に影響しますが、心理的な負担は非常に重いものがあります。
※医療機器規則 (Medical Device Regulation)
※体外診断用医療機器規則 (In Vitro Diagnostics Regulation)
中国国内で実施する生体適合性試験では費用面のメリットが大きいですが、都市部の試験所は近年
試験費用が増加傾向にあります。
弊社では新たな試験委託先の調査を実施しており、この度ISO10993シリーズ/GLP対応の試験所を利用した
試験サービスのご案内が出来るようになりました。

試験所はILAC加盟の中国国家認定もございますので、欧州CEマークやアメリカFDA向けの薬事申請
でも活用いただくことが出来ます。
試験費用は納期や国によっても前後いたしますので、お問合せください。
生体適合性試験のお見積依頼はお気軽にお問い合わせください。
お見積依頼時には医療機器と試験対象となる材料情報、試験項目をご連絡下さい。
香港のお問い合わせをいただく際、「登録は基本任意だから大丈夫!」というお話が多いのですが、
MDCOによる医療機器の規制強制化の動きは進んでいます。
弊社の情報ソースでは2~3年後と予想しております。
強制化後は現行のCAB(Conformity Assessment Body)を活用した制度に近いものとなるようですが、
現時点ではその詳細は明らかになっていません。
来年から!?という噂もございますが、既に上市されている企業様もこれから参入される企業様も早めに
下記の4点について確認を進められることをオススメいたします。
1.移行期間が無い場合を想定して、申請準備から登録までの納期の把握
2.概算費用(コンサル費用、CAB審査費用等)の把握
3.現地代理人のLRP: Local Representative Personのビジネスライセンス有無
4.現地代理人の登録実績・・・申請経験が無いと書類不備等で登録に時間がかかります。
強制化の直前は申請案件で混み合います。
主力商品は強制化前の今から登録してしまうという考え方もあります。
クアルテックは香港におけるLRPとしてMDCOへの製品登録等の実績がございます。
貴社の主導権を持った販売、薬事の仕組み構築を全力でサポートいたします。
弊社ではCFDA向けの現地試験は利用実績の多いCFDAの下部組織となるBIMT:北京市医療器械検験所を利用いたします。
下記は先週金曜のBIMTの施設の写真です。
先週よりお客様の試験を行っており、弊社スタッフは必ず立会います。
試験中は現場とSkypeで逐次状況をご案内させていただくことも可能ですが、
今回のようにお客様に立ち会っていただけるとその場で技術的な問題を議論し解決出来することが出来ます。
試験中はBIMT内のカフェテリアで昼食も取ることが出来ます。


Medical Devices Rules, 2017によると外国の製造者が任命したインド国内の "authorised agent" は輸入業務も行う、となっています。
これはインドネシアと同じ条件のようです。
<英文>
“authorised agent” means a person including any firm or organisation who has been appointed by an overseas manufacturer through a power of attorney to undertake import of medical device in India;
インドで主導権を持って販売・規制対応を進めるためには authorised agentの任命が重要となりそうです。
現地の代理店が輸入者・authorised agentになるケースは一般的ですが、販売の側面では決まった代理店ルートでしか販売出来なくなり拡販の足枷になる可能性があります。
本日、CFDAは外国で承認・許可等を持つ医薬品、医療機器の国内市場へのアクセスを簡易化すると発表しました。
詳細は記載されていませんが、"urgently needed drugs and medical devices" については現行の登録システムの改善に言及しています。
中国市場への製品登録の仕組みがより改善されることを期待いたします。
1.製造ライセンス/販売ライセンス/流通ライセンスはオンライン申請システム採用
Onlince Submission for application for a License to Manufactuer/Sale/Distribute
2.ノーティファイドボディによる監査システム採用
Notified Body audits system
3.外国の製造所はインドのCentral Licensing Authorityによる検査の可能性
Foreign manufacturing sites may be subject to inspection by Central Licensing Authority
4.2020年1月1日以降、機器と製造の識別システムが必須へ
Starting January 1st, 2020, two different types of unique identifies; 1) devce identifier, 2) production identifier
5.アメリカ、EU加盟国、オーストラリア、カナダ、日本の当局による自由販売証明書がある場合、臨床試験は不要
If a CFS has already been issued by the regulatory authority of US, EU, Australia, Canada and Japan, clinical investigation is not required.
6.登録の更新は発行日から5年毎に20,000ルピー(約300USD)と比較的低額に設定
Much less rigorous than that of other countries, regsitration retention fee of 20,0000Rs (approx. 300USD) every five years.
製品規格はインド規格局(Bureau of Indian Standards)が発表しています。
※Medical and Hospital Equipmentsは8つの製品カテゴリに分かれ、合計1121の規格と記載されています。
http://164.100.105.199:8071/php/BIS/IndStndrdLocatr/SubGroupStndrds.php?GId=17#
代表的な電気安全の規格、IEC60601-1は下記のような内容です。
| IS 13450 : Part 1 : 2008/IEC 60601-1 | Medical Electrical Equipment - Part 1 : General Requirements for Basic Safety And Essential Peformance |
また、インドの”Medical Device Rules, 2017”には製品規格について下記のような記載がございます。
<参考訳>
医療機器の製品規格について;
(1)医療機器はBureau of Indian Standardsによって定められた規格又は当局が随時通知する規格に適合すること
(2)(1)に適切な規格が存在しない場合には、適切なISO、IEC又はPharmacopoeial規格に適合すること
(3)(1),(2)に適切な規格が存在しない場合には、製造業者によって検証された規格に適合すること
<原文>
Product standards for medical device.— (1) The medical device shall conform to the standards laid down by the
Bureau of Indian Standards established under section 3 of the Bureau of Indian Standards Act, 1985 (63 of 1985) or
as may be notified by the Ministry of Health and Family Welfare in the Central Government, from time to time.
(2) Where no relevant Standard of any medical device has been laid down under sub-rule (1), such device shall
conform to the standard laid down by the International Organisation for Standardisation (ISO) or the International
Electro Technical Commission (IEC), or by any other pharmacopoeial standards.
(3) In case of the standards which have not been specified under sub-rule (1) and sub-rule (2), the device shall
conform to the validated manufacturer’s standards.
※適用規格の最終判断は、当局への確認をお勧めいたします。
弊社ではインドの製品適用規格の調査が可能ですので、お気軽にお問い合わせください。
調査に必要な情報:製品概要、使用目的と欧州整合規格リスト(英語)